Ritedose verwendete das ChargePoint AseptiSafe® Bio-Ventil mit dem STERIS VHP™-System für den sterilen Pulvertransfer in einem Reinraum der Klasse C und konnte so teure Infrastruktur-Upgrades vermeiden.
Durch einen Vier-Phasen-Zyklus (Entfeuchtung, Konditionierung, Dekontamination und Belüftung) wurde eine validierte Reduzierung um 6 Log-Stufen erreicht, wodurch die Sterilität am Ort der Übertragung gewährleistet wurde.
Die integrierte Lösung machte Isolatoren oder Barrieresysteme mit eingeschränktem Zugang überflüssig, wodurch Kosten, Energieverbrauch und Komplexität reduziert wurden.
Die Validierungsmethoden umfassten chemische und biologische Indikatoren, Medienfüllungen und Untersuchungen zur Sterilitätserhaltung.
Das System verbesserte die Prozesseffizienz, Sterilitätsgarantie und Ergonomie und zeigte damit skalierbare Vorteile für die pharmazeutische Fertigung.
Loading component...
Loading component...
Loading component...
Loading component...
Fallstudie
Zusammenarbeit zwischen STERIS VHP und ChargePoint: Fallstudie Ritedose Corporation
Die Ritedose Corporation ist ein nordamerikanischer Auftragsentwicklungs- und Fertigungsdienstleister (CDMO), der sterile Einzeldosisprodukte herstellt.
2020-10-26
Die Ritedose Corporation ist ein nordamerikanischer Auftragsentwicklungs- und Fertigungsdienstleister (CDMO), der sterile Einzeldosisprodukte herstellt.
Ritedose ist ein Full-Service-Pharmaunternehmen, das die Blow-Fill-Seal-Technologie nutzt. Die Kompetenzen des Unternehmens gehen weit über die Fertigung hinaus: Ein internes Entwicklungsteam ist auf alle Aspekte der Produktmarkteinführung spezialisiert – von Laborchargen über behördliche Zulassungen und die Serienfertigung bis hin zum Vertrieb. Das Unternehmen verfügt über mehr als 20 Jahre Erfahrung in der Herstellung von Atemwegs- und Augenprodukten und verfügt über eine Anlage mit einer Kapazität von 1,7 Milliarden Einheiten, in der die neuesten Technologien in den Bereichen Formulierung, Blow-Fill-Seal und Hochgeschwindigkeitsverpackung zum Einsatz kommen.
Herausforderung
Die Ritedose Corporation suchte nach einer Lösung für das Problem der Befüllung eines Mischbehälters mit einem sterilen pharmazeutischen Wirkstoff (API). Dies ist ein häufig auftretendes Problem bei allen aseptisch hergestellten Arzneimitteln.
Entscheidend für den Prozess war die Aufrechterhaltung steriler Bedingungen beim Andocken eines Behälters an das Gefäß und beim anschließenden Transfer des festen API zur Bildung einer flüssigen Suspension. Bei einer vollständig gelösten Flüssigkeit konnte das Produkt steril gefiltert werden, um die Sterilität beim Weiterleiten an den Füller sicherzustellen. Allerdings handelte es sich in diesem Fall bei dem an den Füller weitergeleiteten Produkt um eine Suspension, sodass diese Option nicht zur Verfügung stand.
Daher musste der gesamte Prozess unter aseptischen Bedingungen durchgeführt werden, was normalerweise bedeutet, dass eines der folgenden Upgrades erforderlich wäre.
1. Aufrüstung des gesamten Raums von einem Reinraum der Klasse C auf Klasse A
2. Aufrüstung des Raums zu einer Umgebung der Klasse B und zusätzliche Einführung eines überdruckbeaufschlagten Bereichs der Klasse A um den Abfüllpunkt herum
3. Aufrüstung des Raums zu einer Umgebung der Klasse B und zusätzliche Einführung eines Barrieresystems mit eingeschränktem Zugang (RABS) am Abfüllpunkt oder am vollständigen Behälter
4. Beibehaltung des Reinraums der Klasse C, aber Einsatz einer Isolatortechnologie um den Abfüllpunkt oder den gesamten Behälter herum
Traditionell würden hier Barrieresysteme mit eingeschränktem Zugang und Isolatortechnologie bevorzugt, da sie durch die grundlegenden Techniken der Trennung und Dekontamination eine verbesserte Sterilitätsgarantie bewirken. Angesichts der negativen Faktoren, die mit diesen Technologien verbunden sind, wie hohe Anfangsinvestitionen, Platzbedarf, Ergonomie sowie laufende Kosten und Energieverbrauch, entschied sich das Unternehmen jedoch, nach einer anderen Lösung zu suchen, die für diese wichtige Aufgabe besser geeignet war.
Lösung
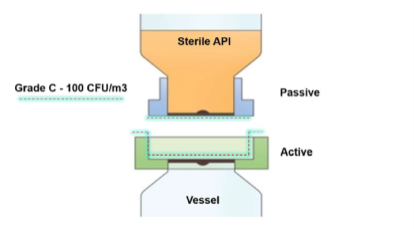
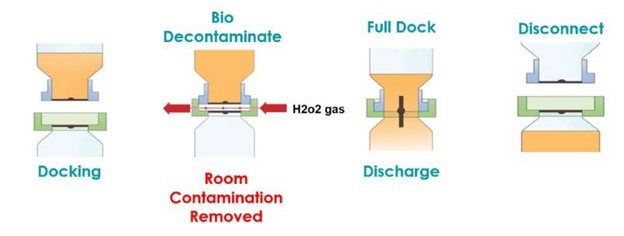
Als ideale Lösung für dieses Problem wurde ein aseptisches Bioventilprodukt ausgewählt, das einen abgedichteten Pulvertransfer auf kleinem Raum ermöglicht und an der Einlassöffnung des Behälters montiert ist. Im Gegensatz zu herkömmlichen Doppelabsperrklappen oder anderen konventionellen Anschlüssen kann das Ventil zusammen mit dem Behälter mit Dampf vorsterilisiert werden (siehe Abbildung 1a/b). Bei der endgültigen Verbindung wurden außerdem alle Verunreinigungen aus dem Raum an den Passflächen des Transfers auf kontrollierte und validierte Weise entfernt (siehe Abbildung 2a/b).
Dock SIP-Kappe Abbildung 1a
SIP durch Kappe – Vorsterilisierung vom aktiven Abschnitt und Gefäß, Abbildung 1b
Steriler API-Transfer
Das AseptiSafe Bio-Ventil funktioniert, indem es eine abgedichtete Kammer zwischen dem Transferbehälter (passiver Abschnitt) und dem Gefäß (aktiver Abschnitt) schafft. Wenn die beiden Hälften zusammengefügt werden, wird die versiegelte Kammer mit einem STERIS-Biodekontaminationssystem mit verdampftem Wasserstoffperoxid (VHP®) biologisch dekontaminiert.
Abbildung 2a
Das STERIS VHP-Gerät entfernt jegliche biologische Verunreinigungen bis zu einer validierten Reduktion um 6 Log-Stufen und hinterlässt den Raum und die Passflächen dekontaminiert und bereit für die vollständige Zusammenführung. Nach dem vollständigen Zusammenfügen kann die Scheibe geöffnet werden, wodurch das Produkt ohne Kontaminationsrisiko vom Transferbehälter in ein Gefäß umgefüllt werden kann. Die Durchführung dieser Übertragung noch im Bereich der Klasse C brachte enorme Kosten- und Produktionsvorteile mit sich, allerdings musste der Prozess vollständig validiert werden, um sicherzustellen, dass die zunächst angenommenen Vorteile auch tatsächlich nachgewiesen werden konnten.
Abbildung 2b
Validierung
Der erste Schritt zur mikrobiologischen Validierung des Prozesses bestand darin, einen validierten Dekontaminationszyklus für die Wasserstoffperoxid-Begasungsphase zu erstellen. Der trockene, gasförmige Prozess von STERIS VHP besteht aus vier verschiedenen Phasen, die der Generator durchläuft, um sicherzustellen, dass alle kritischen Bedingungen erfüllt sind und jedes Mal ein validierter Zyklus durchgeführt wird. Alle vier Phasen sind auf Zeit eingestellt.
1 Entfeuchtung – Das mit Gas beaufschlagte Ventil reduziert die Luftfeuchtigkeit in der Kammer, um ideale Bedingungen für die Abtötung biologischer Keime zu schaffen.
2 Konditionierung – Dabei wird verdampftes Wasserstoffperoxid in das Ventil eingeleitet, bis eine für eine gute Dekontamination ausreichende Konzentration erreicht ist.
3 Dekontamination – Die Konzentration an verdampftem Wasserstoffperoxid wird aufrechterhalten, um jegliche mikrobiologische Aktivität innerhalb des Ventils zu neutralisieren.
4. Belüftung – Nach Abschluss der biologischen Dekontamination wird das verdampfte Wasserstoffperoxid aus dem System entfernt, sodass keine schädlichen Rückstände zurückbleiben. Normalerweise liegt der Akzeptanzwert bei 1 ppm, in diesem Fall wurde jedoch 0,4 ppm als Akzeptanzwert verwendet. Ritedose verwendete einen niedrigeren Rückstandsgrenzwert, um ein zuverlässiges System zu gewährleisten und eine Kontamination des Produkts durch Gasrückstände auszuschließen.
Der vollständige Dekontaminationszyklus kann in nur vier Minuten abgeschlossen werden, normalerweise dauert es jedoch 20 Minuten. Da dieser Prozess nur einmal täglich durchgeführt wurde, wurde zur Gewährleistung eines soliden Zyklus zusätzliche Zeit für jede der kritischen Phasen eingeplant, um sicherzustellen, dass die Dekontamination bestätigt und das Gas aus dem System entlüftet wurde. Dies führte zu einem Gesamtzyklus von 41 Minuten (von der Konditionierung bis zur Belüftung).
Parameter/Phase
Entfeuchtung
Konditionierung
Dekontamination
Aeration
Zeit, Min
10
0
6
25
In den ersten Zyklen wurden chemische Indikatoren (CIs) verwendet, um die H2O2-Verteilung zu bestimmen. Wenn zufriedenstellende CI-Ergebnisse erzielt wurden, wurden biologische Indikatoren (BIs) in den Prozess eingeführt, um zu bestätigen, dass der Prozess erfolgreich durchgeführt wurde. Nach Abschluss jedes Zyklus wurden alle BIs und CIs gesammelt. Anschließend wurden die CI-Streifen auf Farbveränderungen überprüft, um eine gleichmäßige Dampfverteilung sicherzustellen. Die BIs wurden in ein geeignetes Nährmedium, in diesem Fall Spordex® Culture Media, übertragen und sieben Tage lang bei 55 °C bis 60 °C inkubiert. Sie wurden täglich auf mikrobielles Wachstum untersucht.
Zu den Annahmekriterien für den Zyklus gehörten:
A) Alle im Zyklus verwendeten CI-Streifen müssen ihre Farbe geändert haben.
B) Der BI zur Positivkontrolle muss Wachstum aufweisen.
C) Mindestens ein BI von jedem Ort darf kein Wachstum aufweisen.
Nachdem der Zyklus entwickelt worden war, wurde er dreimal durchgeführt, um die Leistungsqualifikation (PQ) für diesen Teil des Prozesses zu bilden.
Um das System vollständig zu validieren, wurde der Prozess vor der Validierung mit mehreren Medienläufen getestet. Diese erfolgreichen Medientests wurden dann mit drei Medienläufen bei PQ fortgesetzt. Die Sterilität wurde nach mehr als 10 Tagen nachgewiesen, wobei das Produkt in das Gefäß überführt wurde und das Bio-Ventil in der geschlossenen, verriegelten Position gehalten wurde. Die Sterilitätserhaltung wurde für den passiven Abschnitt (Produkt im Transferbehälter) für 48 Stunden nachgewiesen, was mehr als ausreichend ist, da diese Zeit in der Regel höchstens halb so lang ist.
Schlussfolgerung
Die Anlage ist nun betriebsbereit und befindet sich in voller Produktion. Zu den ursprünglichen Vorteilen, die zu Beginn des Projekts festgestellt wurden, wie niedrige Investitionskosten, geringer Platzbedarf und einfache Installation, kamen nun auch noch eine verbesserte Sterilitätsgarantie, eine einfache Bedienung und ein geringer Wartungsaufwand hinzu. Das System ist einfach zu verwenden, leicht zu installieren/validieren und hat den Prozess sicherlich verbessert.
Eine Erkenntnis aus diesem Projekt betraf die Dosierungsphase. Zum Zeitpunkt der Validierung war das installierte System eine vollständig starre, wiederverwendbare Lösung, bei der vorsterilisierter API in Beuteln an Ritedose geliefert wurden. Diese Beutel wurden geöffnet und dann aufgeteilt und innerhalb eines aseptischen Isolators in den vorab autoklavierten Transferbehälter und den passiven Abschnitt des Bio-Ventils gegeben. Es wäre vorteilhaft gewesen, Produkt, Behälter und Transferanschluss in einem Schritt (Gammabestrahlung) zu sterilisieren, doch aufgrund der Einschränkungen, die mit der Gamma-Sterilisation von Edelstahl- und Elastomerbaugruppen als ein Gegenstand verbunden sind, war dies nicht möglich.
Diese Lösung ist jetzt von ChargePoint in Form eines Single Use Passive (SUP) / ChargeBag® erhältlich und könnte in Zukunft zur Verbesserung und Rationalisierung des Prozesses eingesetzt werden. Dadurch könnte die gesamte Verpackung (Beutel und Passiv) zur Gammastrahlensterilisation weitergeleitet werden, anstatt mehrere einzelne Sterilisations- und aseptische Montageschritte durchzuführen.
„Es war entscheidend, dass wir für dieses Projekt die richtige Lösung wählten, um Produktkontaminationen und teure Produktverluste zu vermeiden. Wir haben uns für das ChargePoint AseptiSafe® Bio-Transferventil entschieden, weil es eine erhöhte Sterilitätsgarantie bei der Handhabung empfindlicher Inhaltsstoffe wie unserem Arzneimittelwirkstoff bietet. Wir haben während des Projekts umfangreiche Unterstützung erhalten und profitieren von erheblichen Kostensenkungen und Prozessoptimierungen.“
Angie Koen, Vizepräsidentin für technische Dienste, The Ritedose Corporation.
Vorgeschlagene Inhalte
Artikel
Leitfaden zur Implementierung eines VHP-Systems zur Biodekontamination von Einrichtungen
Biodekontaminationssysteme mit verdampftem Wasserstoffperoxid können in eine Vielzahl von Reinräumen oder Anzügen installiert und integriert werden. Hier gibt John Klostermyer, VHP-Anwendungsprojektmanager bei STERIS, einige Tipps für die Erstellung von prozessorientierten Benutzeranforderungsvorgaben.
Auswahl der Dekontaminationstechnologie: Aerosolisiertes vs. verdampftes Wasserstoffperoxid
Dieses Whitepaper untersucht die technischen Aspekte von Wasserstoffperoxid-Aerosolen im Vergleich zur VHP-Technologie, vergleicht deren Leistung anhand wichtiger Kriterien und zeigt zahlreiche Vorteile der VHP-Technologie auf.
Die Vorteile der Dekontamination mit Wasserstoffperoxid-Dampf (VHP)
Informieren Sie sich über die Vorteile der Dekontamination mit Wasserstoffperoxid-Dampf bei Prozessen, bei denen Spuren giftiger Substanzen nicht akzeptabel sind.
Leitfaden zur Implementierung eines VHP-Systems zur Biodekontamination von Einrichtungen
Biodekontaminationssysteme mit verdampftem Wasserstoffperoxid können in eine Vielzahl von Reinräumen oder Anzügen installiert und integriert werden. Hier gibt John Klostermyer, VHP-Anwendungsprojektmanager bei STERIS, einige Tipps für die Erstellung von prozessorientierten Benutzeranforderungsvorgaben.
Auswahl der Dekontaminationstechnologie: Aerosolisiertes vs. verdampftes Wasserstoffperoxid
Dieses Whitepaper untersucht die technischen Aspekte von Wasserstoffperoxid-Aerosolen im Vergleich zur VHP-Technologie, vergleicht deren Leistung anhand wichtiger Kriterien und zeigt zahlreiche Vorteile der VHP-Technologie auf.
Die Vorteile der Dekontamination mit Wasserstoffperoxid-Dampf (VHP)
Informieren Sie sich über die Vorteile der Dekontamination mit Wasserstoffperoxid-Dampf bei Prozessen, bei denen Spuren giftiger Substanzen nicht akzeptabel sind.