Um sicherzustellen, dass das Gerät keine sichtbaren Rückstände aufweist, ist eine Sichtprüfung erforderlich.
„Optisch sauber“ wird akzeptiert, aber die Definitionen variieren, wenn Verfahren begründet sind.
Sowohl direkte als auch indirekte Oberflächen erfordern Hilfsmittel für eine effektive Sichtprüfung.
Validierte Methoden müssen eine Sichtprüfung unterstützen und so eine gründliche Sauberkeit gewährleisten.
Sichtbarkeitsschwellenwerte definieren die Kontaminationserkennung für eine konsistente Inspektion.
Loading component...
Loading component...
Loading component...
Loading component...
Artikel
Verfahren zur Sichtprüfung gereinigter Geräte
Gemäß den gesetzlichen und Arzneibuchrichtlinien müssen Hersteller bestätigen, dass die Prozessausrüstung nach einem Reinigungsvorgang optisch sauber ist. Eine kürzlich durchgeführte Umfrage von Steris ergab, dass die Verfahren zur Sichtprüfung gereinigter Geräte je nach Hersteller unterschiedlich sind. Gleichzeitig zeigte die Umfrage, dass zwar die Praktiken und sogar die Begrifflichkeiten unterschiedlich sein können, dies jedoch von den Aufsichtsbehörden akzeptiert wird, sofern die Prozesse gut dokumentiert sind.
2020-05-12
Gemäß den gesetzlichen und Arzneibuchrichtlinien müssen Hersteller bestätigen, dass die Prozessausrüstung nach einem Reinigungsvorgang optisch sauber ist. Eine kürzlich durchgeführte Umfrage von Steris ergab, dass die Verfahren zur Sichtprüfung gereinigter Geräte je nach Hersteller unterschiedlich sind. Gleichzeitig zeigte die Umfrage, dass zwar die Praktiken und sogar die Begrifflichkeiten unterschiedlich sein können, dies jedoch von den Aufsichtsbehörden akzeptiert wird, sofern die Prozesse gut dokumentiert sind.
Zunächst ein Wort zu den Begrifflichkeiten: In vielen behördlichen Richtlinien und technischen Dokumenten der Branche werden unterschiedliche Begriffe verwendet, um den Schritt zu beschreiben, mit dem bestätigt wird, dass die Prozessausrüstung nach der Reinigung optisch sauber ist. Für die Zwecke dieses Artikels verwenden die Autoren den Begriff „Sichtprüfung“. Andere gebräuchliche Begriffe für diesen Schritt sind „visuelle Inspektion“ oder „Sichtkontrolle“. Beachten Sie, dass sich „Sichtprüfung“ in diesem Fall nicht auf die visuelle Überprüfung des Endprodukts auf Partikel bezieht.
An der Umfrage nahmen 39 Personen teil, die viele europäische Pharma- und Biopharmaunternehmen repräsentieren (27 Unternehmen an 34 verschiedenen Standorten). An der Umfrage nahmen Hersteller aus folgenden Bereichen teil: 54 % nicht sterile Produkte (etwa Tabletten, Flüssigkeiten, Kombinationsprodukte), 13 % sterile Produkte (etwa Biotechnologie, Flüssigkeiten und lyophilisierte Produkte), 26 % Impfstoffe, 5 % Medizinprodukte, 2 % Sonstiges (etwa frühe klinische Produktionsphasen).
Der EU-GMP-Anhang 15 besagt, dass „eine Sichtprüfung der Reinigung ein wichtiger Bestandteil der Akzeptanzkriterien für die Reinigungsvalidierung ist“ (1). Die Sichtprüfung ist ein wichtiger Schritt, um die Wirksamkeit der Reinigungsprozessausrüstung nach der Reinigung zu bestätigen. Das Abnahmekriterium für die Sichtprüfung ist optische Sauberkeit. Die Sichtprüfung sollte direkte und indirekte Produktkontaktflächen umfassen und erfordert, dass die Geräteoberflächen sichtbar sind. Ist dies nicht der Fall, kann es erforderlich sein, die Ausstattungen zu demontieren, um Zugang zu erhalten, oder Hilfsmittel wie Spiegel, Lichtquellen oder Endoskope zu verwenden (2, 3). Moderne Technologien wie Digitalkameras, die die Oberfläche beurteilen können, könnten auch für großvolumige Behälter in Betracht gezogen werden, wenn eine Sichtprüfung schwierig ist.
Optische Sauberkeit ist der erwartete Mindeststandard. Es wird jedoch ein zusätzliches Akzeptanzkriterium, wie der gesundheitsbezogene Grenzwert, durchgesetzt (1-5). Daher sollte eine validierte Analysemethode mit einer Empfindlichkeit unterhalb der Reinigungsgrenze regelmäßig mit einer Sichtprüfung kombiniert werden. Wenn die Sauberkeit von Geräten nur durch Sichtprüfung bestimmt wird, sollte ein Grenzwert festgelegt werden, bei dem das Produkt als Rückstand deutlich sichtbar ist (3, 6).
Die Sichtprüfung wird (soweit möglich) immer am Ende eines vollständigen Reinigungszyklus durchgeführt (7). Bei der Sichtprüfung handelt es sich um eine aktive und qualitative Beobachtung der Produktkontaktflächen, um die Nichtvorhandensein von Rückständen zu bestätigen, damit mit der Produktion der nächsten Charge begonnen werden kann (7). ICH Q7: In den Praxisleitlinien für pharmazeutische Wirkstoffe heißt es: „12.76 … Durch eine Sichtprüfung können grobe Verunreinigungen erkannt werden, die sich auf kleine Bereiche konzentrieren und bei der Probenahme und/oder Analyse sonst unentdeckt bleiben könnten.“
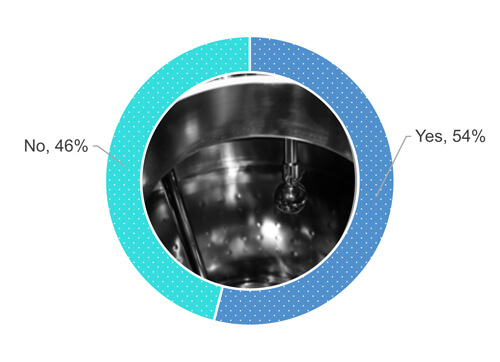
Zurück zur Umfrage: 54 % der Befragten führen eine Sichtprüfung der Ausstattungsoberflächen durch, wenn diese trocken ist (Abbildung 1).
Abbildung 1: Verlangen Ihre Unternehmensabläufe, dass Sie nach einer Reinigung eine Sichtprüfung der Oberflächen der Prozessausrüstung durchführen, wenn diese trocken sind?
Einige technische Dokumente der Branche empfehlen, die Sichtprüfung nach Möglichkeit auf einer getrockneten Oberfläche durchzuführen, um falsch-negative Ergebnisse zu vermeiden (8, 9):
„Nach der Durchführung der Reinigungsvorgänge sollte die Ausrüstung getrocknet werden, um eine Sichtprüfung zu ermöglichen.“
„Das Abnahmekriterium für die Reinigung von Ausrüstung sollte auf optischer Sauberkeit in trockenem Zustand und einem analytischen Grenzwert basieren.“
PDA Technical Report Nr. 29: Zu berücksichtigende Punkte bei der Reinigungsvalidierung (9): „Normalerweise sollten Oberflächen, die einer Sichtprüfung unterzogen werden, trocken sein, da dies die widrigsten Bedingungen für eine Sichtprüfung darstellt.
Es ist allgemein bekannt, dass bei einigen Rückständen eine optisch saubere Oberfläche nur erreicht werden kann, wenn die Oberfläche nass ist, während dies nach dem Trocknen nicht mehr der Fall ist.
Die Konstruktion der Anlage und die Parameter des Reinigungszyklus können dazu führen, dass die Oberflächen der Anlage trocknen, etwa wenn die Leitungen zum Abfluss hin geneigt sind und der Abfluss selbstständig erfolgt, und die Endspülung bei hohen Temperaturen durchgeführt wird. In einigen Fällen kann das Einblasen sauberer Luft in die Geräte und das Verteilungssystem dazu beitragen, trockene Oberflächen zu erreichen (9).
Obwohl die oben aufgeführten Parameter erfüllt sind, können dennoch Tröpfchen oder Feuchtigkeit („Schwitznässe“) auf der Oberfläche auftreten, die jedoch bei angemessener Begründung akzeptabel sein können.
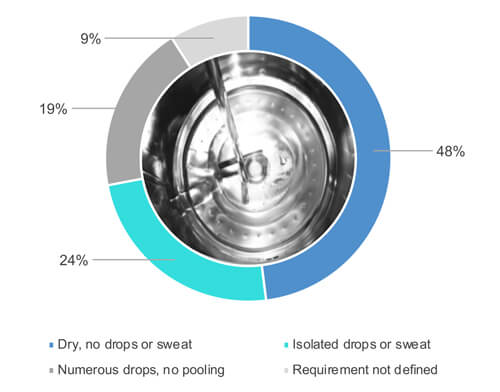
Wie Abbildung 1 zeigt, antworteten 48 % von 54 % mit „Ja“, als sie gefragt wurden, ob sie die Oberflächen von Geräten einer Sichtprüfung unterziehen, wenn sie vollständig trocken sind (Abbildung 2). 24 % der 54 % lassen bei einer Sichtprüfung der gereinigten Ausrüstung eine gewisse partielle Nässe (etwa vereinzelte Tropfen oder Schwitznässe) der Oberflächen zu (Abbildung 2). 19 % der 54 % akzeptieren zahlreiche Tropfen, aber keine Wasseransammlungen. 9 % der 54 % haben keine Anforderungen hinsichtlich der Trockenheit festgelegt.
Abbildung 2 Welche Trockenheitsanforderungen gelten für die Prozessausrüstung nach Abschluss des Reinigungszyklus?
Unterschiedliche Trockenheitsgrade sind akzeptabel, wenn sie ausreichend begründet und durch Daten gestützt sind, die belegen, dass keine Auswirkungen auf die Sichtprüfung nach der Reinigung und die Vermehrung der Keimbelastung während der sauberen Lagerung vorliegen, etwa durch strenge Anweisungen und Schulungen unter Verwendung von Fotos, um Abweichungen zu vermeiden. Darüber hinaus erlaubt keiner der 54 % der Hersteller, die ihre Geräte bei trockenen Oberflächen einer Sichtprüfung unterziehen, dass Wasser auf den Geräteoberflächen verbleibt oder sich dort ansammelt, wie in den US-amerikanischen Richtlinien empfohlen. FDA-Inspektionsleitfaden (10): „… Beispielsweise sollten Geräte vor der Lagerung getrocknet werden, und nach der Reinigung darf auf keinen Fall Restwasser in den Geräten zurückbleiben.“
Es liegt in der Verantwortung des Bedieners, zu entscheiden, ob die Oberflächen der Geräte optisch sauber sind. Daher sollten das Bedienpersonal alle Oberflächen der Geräte einer Sichtprüfung unterziehen können. Ist dies nicht möglich, sollten geeignete oder fortschrittliche Instrumente zur Verfügung stehen, um eine ordnungsgemäße Entscheidungsfindung zu gewährleisten (2, 3, 9, 11). Eine Sichtprüfung eines großen Behälters durch ein Schauglas ist aufgrund der verdeckten Oberfläche nur eingeschränkt möglich. Die EMA schlägt vor, dass die Möglichkeit der Sichtprüfung der Ausrüstung, wie beispielsweise die vor Ort festgestellten Abstände, in einem Frage-und-Antwort-Dokument berücksichtigt werden sollte (3), und empfiehlt, dass „schriftliche Anweisungen, in denen alle Bereiche angegeben sind, die einer Sichtprüfung unterzogen werden müssen, vorhanden sein sollten und dass aus den Aufzeichnungen eindeutig hervorgeht, dass alle Überprüfungen vollständig durchgeführt wurden“. Schließlich sind detaillierte Verfahren und Schulungen hinsichtlich des Kriteriums der optischen Sauberkeit zwingend erforderlich, um eine korrekte Entscheidungsfindung zu gewährleisten. Das Niveau der Schulung und Qualifikation für die Sichtprüfung sollte dem Risiko einer Kreuzkontamination angemessen sein (3, 7, 9, 11).
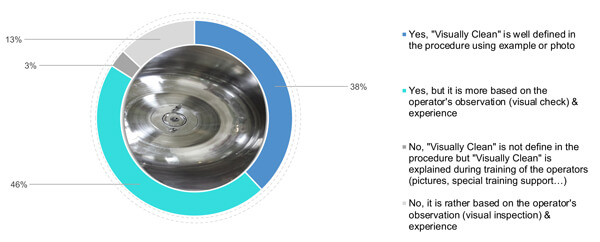
Was Fotos betrifft, haben 38 % der Befragten das Kriterium der optischen Sauberkeit zusätzlich durch Fotos und Beispiele für Sauberkeit näher definiert (Abbildung 3). 46 % verlassen sich auf die Einschätzung des Bedieners in Bezug auf „visuell sauber“ in Verbindung mit einer theoretischen Definition von „visuell sauber“ im Verfahren. Schließlich verlassen sich 16 % (3 % und 13 %) auf die Erfahrung und Schulung des Bedienpersonals (unter Verwendung spezifischer Hilfsmittel oder Bilder) hinsichtlich einer optisch sauberen Oberfläche (Abbildung 3).
Abbildung 3: Definiert und erläutert Ihr Unternehmen die Bedeutung des Kriteriums „optische Sauberkeit“?
Bedienpersonal, das Sichtprüfungen durchführt, muss speziell geschult sein, da das, was man sehen kann, je nach Entfernung, Winkel, Beleuchtung, Beschaffenheit der Oberfläche, Trockenheitsgrad und Sehschärfe des Prüfers variiert (3, 7, 9). Die EMA empfiehlt, regelmäßig Sehtests (oder Sehschärfetests) durchzuführen und die Kompetenz des Bedienpersonals durch eine praktische Prüfung nachzuweisen (3). Die ISPE Risk Mapp schlägt Folgendes vor: „In Situationen, in denen die Erkennung nur visuell erfolgt, ist es wichtig, die Sehschärfe des Personals zu kennen und zu wissen, welcher Grad an Rückständen als sicher gilt (7). Wenn das Sicherheitsniveau unter der Sehschärfe des Personals liegt, kann das Risiko, dass Mängel nicht erkannt werden, als hoch angesehen werden, während das Risiko, dass Mängel nicht erkannt werden, als gering angesehen werden kann, wenn das Sicherheitsniveau deutlich über (um ein Vielfaches) der Sehschärfe des Personals liegt.
Die Häufigkeit der Sehtests und die Sehschärfegrenze hängen von bestimmten Einflussfaktoren ab, wie zum Beispiel:
Abstand zwischen dem Prüfer und der zu prüfenden Geräteoberfläche (3, 7, 12)
Gerätekonfiguration und Oberflächenbeschaffenheit (7, 8)
Toxizität von Rückständen, Reinigungsgrenze gegenüber visueller Nachweisgrenze (3, 6, 7)
Eine Sichtprüfung wird nur durchgeführt, um die Sauberkeit einer Ausrüstung zu bestätigen (7)
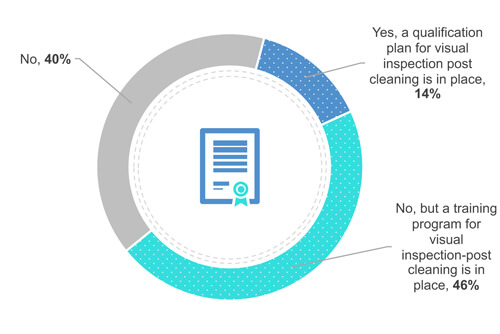
38 % der Befragten verlangen keine Zertifizierung oder Qualifizierung des Bedienpersonals. (Abbildung 4). Dennoch schulen 49 % ihr Bedienpersonal darin, die Geräte vor Ort (vor der Ausrüstung) optisch sauber zu halten. Schließlich qualifizieren 13 % ihr Bedienpersonal.
Abbildung 4: Verfügt Ihr Unternehmen über ein Zertifizierungs- oder Qualifizierungsprogramm für die Sichtprüfung?
Einige Hersteller haben den Grenzwert für sichtbare Rückstände anhand von Teststreifen definiert, die das Bedienpersonal erkennen konnte (13, 14). Einige ahmten auch den Abstand zwischen dem Bedienpersonal und den zu prüfenden Oberflächen an den Prozessausrüstungen nach (15, 16). Wenn die Grenze für sichtbare Rückstände unterhalb der Reinigungsgrenze liegt, kann eine Sichtprüfung als Abnahmekriterium ausreichen (7).
Gewährleistet eine Zertifizierung oder Qualifizierung in Verbindung mit einem Schulungsprogramm die Kompetenz des Bedienpersonals zur Durchführung einer Sichtprüfung der Oberfläche? Gemäß dem Grundsatz von ICH Q9: Qualitätsrisikomanagement (17), hängt die Antwort im Allgemeinen von folgenden Faktoren ab:
Anzahl der bisherigen Abweichungen bei der Sichtprüfung bedingt durch die Schulung des Bedienpersonals
Schwierigkeiten bei der Überprüfung der Ausrüstung (Ausrüstungskonfiguration und Umgebungsbedingungen wie Beleuchtung, Winkel usw.) (3, 7–9, 11, 12, 18, 19)
Art der durchgeführten Sichtprüfung; qualitative oder quantitative Sichtprüfung (7,12, 13–16, 18,19)
Die Sichtprüfung wird nicht durch analytische Prüfungen ergänzt.(13–16,18)
Häufigkeit der mit Sichtprüfung durchgeführten analytischen Tests
Empfindlichkeit der Analysemethode gegenüber der Reinigungsgrenze, Toxizität (basierend auf dem gesundheitsbezogenen Expositionsgrenzwert) der Rückstände (1–7, 9, 11)
Vorhandensein einer doppelten Überprüfung
Schlussfolgerung
Das Konzept der Sichtprüfung und das Kriterium der „optischen Sauberkeit“ scheinen je nach Erfahrung mit der Durchführung des Reinigungsprozesses und dem Verständnis der gesetzlichen Anforderungen bei den europäischen Herstellern unterschiedlich zu sein. Das Kriterium der optischen Sauberkeit muss jedoch in den Verfahren klar definiert sein. Bedienpersonal, das Sichtprüfungen durchführt, muss speziell geschult sein, wobei diese Schulung auf den eigenen Erfahrungen aufbauen kann.
Die in den Umfrageergebnissen aufgezeigten Unterschiede in den Praktiken der Hersteller können akzeptabel sein, wenn das Risiko dokumentiert ist. Gemäß den Vorgaben der ICH Q9 sollten der Aufwand und die Formalität in einem angemessenen Verhältnis zum Risiko für den Patienten stehen.
In Teil II werden eine Fallstudie und Mindestanforderungen für die Sichtprüfung gereinigter Oberflächen vorgestellt.
Teil II
Europäische Hersteller interpretieren das Kriterium „optisch sauber“ aufgrund ihrer Erfahrungen mit der Durchführung des Reinigungsprozesses und ihrer eigenen Auslegung der gesetzlichen Anforderungen unterschiedlich (1). Dennoch sollten ein formelles Schulungs- und Qualifizierungsprogramm für die Sichtprüfung sowie entsprechende Anweisungen eingeführt werden, um das Kontaminationsrisiko zu vermeiden und sicherzustellen, dass die Geräte gemäß den Vorgaben des Herstellers gereinigt werden (2).
Unterschiede in der Vorgehensweise der einzelnen Hersteller sind akzeptabel. Das Bedienpersonal muss jedoch regelmäßig darin geschult und qualifiziert werden, wann die optische Sauberkeit der Ausrüstung zu überprüfen ist und wie zwischen Produktrückständen und Oberflächenfehlern, die die optische Sauberkeit beeinträchtigen können, unterschieden werden kann. Die folgende Fallstudie veranschaulicht, warum dies wichtig ist.
Fallstudie: Inspektionsbeobachtung bei Sichtprüfung
Kürzlich haben Inspektoren der US-amerikanischen FDA einem nicht namentlich genannten biopharmazeutischen Hersteller eine Beobachtung hinsichtlich der Kriterien für optische Sauberkeit mitgeteilt. Bei einem Rundgang durch die Produktionshalle des Unternehmens besichtigte einer der Inspektoren den vorgelagerten Raum, in dem sich zwei Fermenterbehälter befanden. Die Behälter sind Teil eines geschlossenen Systems und werden mit einem vollautomatischen Clean-in-Place-System (CIP) gereinigt. Der CIP-Zyklus eines der Fermenter wurde eine Stunde vor Betreten des vorgelagerten Raums durch den Inspektor abgeschlossen. Der Prüfer blickte durch das Schauglas in das Innere des Behälters, beobachtete einige Kondensationstropfen und stellte fest, dass die Oberflächen nicht glänzten. Dies überzeugte den Prüfer nicht davon, dass der Behälter „optisch sauber“ war (Abbildung 1).
Abbildung 1 Einige chemische Fingerabdrücke (Verfärbungsspuren) wurden auf der Innenfläche des Fermenters festgestellt.
Dies führte zu folgender Beobachtung: „Im Fermenter (Geräte-ID XXXX) wurde eine Verfärbung beobachtet. Dieses spezielle Problem wurde von der Firma nicht bewertet. Dieser Fermenter wird für die Fermentation von [Produktname] verwendet.“ [Anmerkung des Autors: Die Zitate stammen aus dem daraus resultierenden Dokument der Aufsichtsbehörde.]
Diese Beobachtung warf viele Fragen auf und hatte Auswirkungen auf mehrere Kontrollstrategien. Da es nach einem Reinigungszyklus auftrat, bei dem eine systematische (routinemäßige) Sichtprüfung durchgeführt wurde, hatte dies Auswirkungen auf die Reinigungssteuerungsstrategie und das Wartungsprogramm, mit denen sichergestellt werden sollte, dass die Behälteroberflächen in einem akzeptablen Zustand waren. Diese beiden Programme waren hinsichtlich der Inspektion und Handhabung von Behältern nicht aufeinander abgestimmt. Was könnte praktischer sein als eine tägliche Sichtprüfung, um einen akzeptablen Oberflächenzustand zu bestätigen, bevor der Status „optisch sauber“ bestätigt wird?
Das biopharmazeutische Unternehmen konnte das Vorhandensein von Tröpfchen nach Abschluss des CIP-Prozesses als akzeptabel rechtfertigen, hauptsächlich weil die Zeit im gereinigten Zustand (Clean Hold-Time, CHT) unter ähnlichen Sauberkeitsbedingungen erfolgreich validiert worden war. Der Prüfer verstand die Situation (dass die Ausrüstung unmittelbar nach dem CIP-Prozess begutachtet wurde) und akzeptierte die Begründung.
Auch zu den Verfärbungen an der Behälterwand gab es eine Erklärung. Verschiedene Bedingungen können zu einer Verfärbung des Edelstahls führen, wenn dieser mit bestimmten Produkten, chemischen Mitteln oder thermischen Einflüssen in Berührung kommt. Dies wird als oberflächliche interne Verwitterung bezeichnet. Durch die Veränderung der Oberfläche können sich die Bedingungen für die Sichtprüfung ändern, was wiederum die Entscheidung des Prüfers (oder des Bedienpersonals) darüber beeinflussen kann, ob die Oberfläche optisch sauber ist – selbst wenn der Hersteller über einen hochwertigen Schulungs- und Qualifizierungsprozess verfügt.
Die Beobachtung selbst ist eindeutig: Die „Verfärbung … wurde nicht bewertet“, während die Ausrüstung routinemäßig zur Herstellung biologischer Massenprodukte verwendet wurde. Es wurde jedoch nicht auf eine bestimmte Anforderung Bezug genommen.
Wie hat das Unternehmen auf diese Beobachtung reagiert?
Das Unternehmen räumte ein, dass bei der FDA-Inspektion eine Oberflächenverfärbung festgestellt wurde, die vor der Inspektion nicht bewertet worden war.
Es wurden umgehend Maßnahmen zur Untersuchung der Verfärbung ergriffen, Proben zur Analyse entnommen und der Fermenter nicht verwendet.
Die Untersuchung ergab, dass die Dekontamination der Behälter mit ätzenden oder anderen chemischen Mitteln bei hohen Temperaturen (über 100 °C) die Behälterwände belastete und eine chemische Reaktion auf Edelstahloberflächen auslösen konnte. Das Elektropolieren stellt die Oberfläche der Gefäße nur vorübergehend wieder her. Tatsächlich haben die Dekontaminations- und Reinigungsprozesse seitdem zu Verfärbungen auf den Innenflächen des Fermenters geführt.
Aus dieser Beobachtung der FDA lässt sich eine Lehre ziehen: Der Umfang der Sichtprüfung durch einen Prüfer (beispielsweise Bedienpersonal oder technisches Personal) nach einem Reinigungsvorgang sollte klar definiert sein. Die Dekontaminierungs- und Reinigungsprozesse haben im Laufe der Zeit einen nachhaltigen Einfluss auf die Struktur der Ausrüstung. Die vorhandenen Prozesse zur Reinigung, Dekontaminierung und Wartung müssen konsistent, aufeinander abgestimmt und schrittweise erfolgen. Daher muss die Reinigungs- und Wartungskontrollstrategie in der Lage sein, Antworten auf folgende Probleme zu liefern:
Wie bewertet und dokumentiert man die „optischen Mängel“ des Materials, aus dem die Ausrüstung besteht?
Wie lässt sich ein akzeptabler Trocknungsgrad vor der Sichtprüfung angemessen definieren?
Wie schult man Inspektoren (Bedienpersonal, Techniker und Fachexperten) in Bezug auf optisch saubere Oberflächen und akzeptable Oberflächenfehler?
Wie definiert man einen Oberflächenfehler, der zur Disqualifizierung der Ausrüstung führen würde?
Wie lässt sich feststellen, ob die Sichtprüfung auch die Kontrolle von Oberflächenfehlern umfassen sollte?
Überprüfung des optisch sauberen Zustands
Die meisten Prüfer (Bedienpersonal) in der gesamten Branche haben Fertigungsausrüstung auf der Grundlage ihrer Erfahrung geprüft und freigegeben (1,2). Allerdings kann es für das Bedienpersonal schwierig sein, Produktrückstände und Oberflächenfehler zu unterscheiden.
Um dieses Problem zu beheben, sollte das Bedienpersonal an Ausrüstung geschult werden, die aus denselben Materialien besteht und dieselben Eigenschaften aufweist wie das Material, bei dem die Rückstände visuell erkannt werden sollen. Das Schulungsverfahren und die Unterstützung zur Gewährleistung einer reproduzierbaren Sichtprüfung sollten mindestens die folgenden Abschnitte enthalten:
1 Definition des Kriteriums der optischen Sauberkeit
Optisch sauber, definiert als „das Nichtvorhandensein sichtbarer Rückstände auf einer Oberfläche“, sollte in dem Verfahren ordnungsgemäß als Abnahmekriterium definiert werden.
Neben Beschreibungen sind Bilder (Abbildung 2) von optisch sauberen Oberflächen in einem Verfahren oder einer Schulung die beste Option. Durch die Sichtprüfung konnten weit mehr als nur Produktrückstände festgestellt werden.
Jüngste Studien haben gezeigt, dass es für die Sichtprüfung sauberer Oberflächen wichtig ist, den Prüfer darin zu schulen, zwischen Rückständen auf der Oberfläche und unkritischen Oberflächenfehlern oder Verfärbungen zu unterscheiden (2, 3). In einigen Fällen könnte das Bedienpersonal hinsichtlich des zulässigen Ausmaßes an Verfärbungen oder Mängeln auf den Oberflächen informiert werden, da einige diese Oberflächenkontrolle als Teil des Wartungs- oder Instandhaltungsprogramms betrachten.
Der Grad der akzeptablen Trockenheit von Geräten oder die Kategorie und Bedingungen der Abflussfähigkeit könnten in die Definition aufgenommen oder in eine Reinigungsrisikobewertung einbezogen werden.
Abbildung 2 Optisch saubere Oberflächen nach einem Reinigungsvorgang
2 Umfang der Sichtprüfung
Welche Oberflächen müssen einer Sichtprüfung unterzogen werden? Alle Oberflächen der Geräte, die direkt oder indirekt mit dem Produkt, dem Chargen- oder Zwischenmaterial in Berührung kommen, sind einer Sichtprüfung zu unterziehen. Da bei einer Sichtprüfung möglicherweise nicht alle Oberflächen eines Geräts sichtbar sind, kann der Umfang auf die Art des Geräts zugeschnitten werden. Es sollte besprochen werden, wie und was an der Oberfläche visuell zu überprüfen ist.
3 Umgebungsbedingungen zur visuellen Prüfung einer sauberen Oberfläche
Die Bedingungen in der Umgebung der zu überprüfenden Ausrüstung beeinflussen die Fähigkeit, Rückstände ordnungsgemäß zu erkennen, sodass die Umgebung ein wichtiger Faktor ist. Die Menge der nachweisbaren Rückstände (Grenzwert für sichtbare Rückstände) ist produktabhängig und muss von Fall zu Fall oder anhand des Gruppierungsansatzes (2–6) festgelegt werden.
Um den Einfluss der Umgebungsbedingungen auf eine Sichtprüfung zu bestätigen, sollten die folgenden Faktoren analysiert werden:
Betrachtungsabstand: Der Abstand zwischen der Oberfläche und dem Bedienpersonal. Dieser Betrachtungsabstand lässt sich in einem Labor möglicherweise nicht einfach simulieren, allerdings ist nach gesundem Menschenverstand davon auszugehen, dass die visuelle Prüfung umso schwieriger ist, je größer der Abstand ist. Auch die Sehschärfe des Prüfers auf eine bestimmte Entfernung spielt eine Rolle.
Lichtstärke (ca. 400–1500 Lux in normalen Reinräumen): Einige Untersuchungen mit Teststreifen haben gezeigt, dass Lichtstärken zwischen 200 und 1400 Lux die visuelle Inspektion eines Prüfers nicht beeinträchtigen. Dies muss jedoch noch unter realen Bedingungen nachgewiesen werden.
Betrachtungswinkel: Auch der Blickwinkel zwischen den Augen des Prüfers und den visuell zu prüfenden Oberflächen sollte berücksichtigt und in den Prozess der visuellen Inspektion integriert werden. Je nach Winkel und Lichtstärke kann ein Prüfer Reflexionen wahrnehmen, die eine visuelle Sauberkeit erschweren.
Sekundärlicht: Die Verwendung einer Taschenlampe wird nicht empfohlen, wenn die Reinigungsvalidierungsläufe nicht mit einer Taschenlampe überprüft wurden. Die Bedingungen für die Inspektion müssen bei Validierungsläufen und bei der routinemäßigen Reinigung und Wartung ähnlich sein.
Das Verständnis der Umgebungsfaktoren, die die Sichtprüfung beeinflussen können, ist entscheidend, um Fehlentscheidungen zu vermeiden.
4 Zeitpunkt der Durchführung der visuellen Inspektion
Eine Sichtprüfung sollte nach Abschluss des Reinigungsvorgangs durchgeführt werden. Gleichzeitig ist dies oft nicht Teil der Dokumentation und kann zu Fragen über die Durchführung der Sichtprüfung führen, die im Falle eines abgebrochenen Reinigungszyklus durchgeführt wird. Das Verfahren sollte festlegen, wie lang die Zeitspanne nach dem Ende des Reinigungszyklus sein sollte, bevor eine Sichtprüfung durchgeführt wird. Dieser Zeitpunkt sollte so festgelegt werden, dass die Sicherheit des Bedienpersonals beim Öffnen eines Behälters gewährleistet ist (beispielsweise wird der Spülschritt in der Regel bei hohen Temperaturen durchgeführt). Dieser Zeitpunkt wirkt sich auf den Wasser- oder Lösungsmittelablass aus und könnte die Inspektion vorzeitig ungültig machen.
5 Methodik zur Durchführung der Sichtprüfung
Es sollte eine strukturierte Methodik zur visuellen Überprüfung sauberer Geräte festgelegt werden. Ein Hersteller könnte beispielsweise eine Checkliste erstellen, in der die verschiedenen Teile der Ausrüstung aufgeführt sind, die in einem zusammenhängenden visuellen Pfad einer Sichtprüfung unterzogen werden müssen. Zusätzlich zur Methode für die allgemeine Sichtprüfung haben einige Hersteller Checklisten erstellt, in denen bestimmte Teile oder bekannte problembehaftete Stellen aufgeführt sind, die einer Sichtprüfung unterzogen werden sollten.
6 Identifizierung der sichtbaren Zielrückstände
Das Bedienpersonal muss mit allen Elementen vertraut gemacht werden, die es bei der Sichtprüfung überprüfen kann. Zu diesen Elementen können unter anderem Produktrückstände, Oberflächenfehler, Oberflächenschäden, Restwasser und Partikel gehören.
Das Bedienpersonal überprüft die sichtbaren Oberflächen auf alle Mängel. Sie werden auch viele andere Elemente oder Anomalien erkennen können, angefangen bei Materialfehlern, Alterungserscheinungen, Spuren von Restwasser, Partikeln und Produktrückständen (Abbildung 3). Daher müssen das Ziel der Sichtprüfung und die Abnahmekriterien klar definiert sein.
Abbildung 3 Elemente, die das Bedienpersonal an einer direkten oder indirekten Produktkontaktfläche erkennen muss
Der Grad der formalen Ausgestaltung dieser sechs Abschnitte hängt von der Risikobewertung und den Kontrollstrategien des Unternehmens hinsichtlich der Reinigung ab.
Schlussfolgerung
Zur Bestätigung eines optisch sauberen Zustands ist eine Sichtprüfung die Methode der Wahl. Die meisten Prüfer (Bedienpersonal) in der gesamten Branche prüfen und geben Fertigungsausrüstung auf der Grundlage ihrer Erfahrung frei (1,3). Für das Bedienpersonal könnte es jedoch schwierig sein, ohne entsprechende Schulung und Qualifizierung Produktrückstände und Oberflächenfehler zu unterscheiden. Daher ist es sinnvoll, das Bedienpersonal entsprechend zu schulen und sicherzustellen, dass alle Konstruktionsmaterialien, und zwar unter ähnlichen Bedingungen, bereitgestellt werden, um Zielrückstände und Oberflächenfehler, die den optisch sauberen Zustand beeinträchtigen könnten, visuell zu erkennen. Es sollten ein formelles Schulungs- und Qualifizierungsprogramm für die Sichtprüfung sowie entsprechende Anweisungen erstellt werden. Dieses Programm sollte auf den Kontrollen basieren, die der Hersteller verwendet, um sicherzustellen, dass die Ausrüstung sauber ist. Daher könnte die definierte und qualifizierte Sichtprüfung eine erste Kontrollmethode im Rahmen einer Strategie zum Reinigungsrisikomanagement sein.
Beide Autoren waren an der Erstellung des Manuskripts beteiligt und haben die endgültige Fassung genehmigt. Die Erstellung dieses Artikels wurde von GlaxoSmithKline Biologicals SA und STERIS gesponsert. Die Autoren erklären folgendes Interesse: Walid El Azab ist Mitarbeiter von STERIS, Stephane Cousin ist Mitarbeiter der GSK-Unternehmensgruppe.
Verweise Teil 1
European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use: Annex 15, qualification and validation, 2015.
Canada Health Products and Food Branch Inspectorate. Guidance Document. Cleaning validation guidelines: Drug and health products. Health Canada: Ottawa, Canada; 2002 Spring.
European Medicines Agency, Questions and answers on implementation of risk-based prevention of cross-contamination in production and ‘Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities’ (EMA/CHMP/CVMP/SWP/169430/2012), EMA/CHMP/CVMP/SWP/246844/2018, (April 2018)
European GMP part IV Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products
Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme. Guide to Good Manufacturing Practice for Medicinal Products: Annex 15, 2015.
Walsh A. et al., “Justification & Qualification Of Visual Inspection For Cleaning Validation In A Low-Risk, Multiproduct Facility.” Pharmaceutical Online (Aug. 3, 2018) accessed January 2020 https://www.pharmaceuticalonline.com/doc/justification-qualification-of-visual-inspection-for-cleaning-validation-in-a-low-risk-multiproduct-facility-0001
ISPE, ISPE Risk-Based Manufacture of Pharmaceutical Products, second edition, Volume 7
Active Pharmaceutical Ingredients Committee, guidance on aspects of cleaning validation in active pharmaceutical ingredient plants (2016)
Parenteral Drug Association, Technical Report 29, Points to Consider for Cleaning Validation (2012)
GUIDE TO INSPECTIONS VALIDATION OF CLEANING PROCESSES, accessed on September 2019: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-guides/validation-cleaning-processes-793
Parenteral Drug Association, Technical Report 49, Points to Consider for Biotechnology Cleaning Validation (2010)
ASTM: E306-18, Standard guide for Science-Based and Risk-Based Cleaning Process Development and Validation, October 2018 version 1.
Forsyth, R.J., et al. “Visible-Residue Limit for Cleaning Validation and its Potential Application in a Pharmaceutical Research Facility.” Pharmaceutical Technology 28 (Oct. 1, 2004) 68–72.
“Application of Visible-Residue Limit for Cleaning Validation.” Pharmaceutical Technology 28 (Oct. 2, 2005) 10 http://www.pharmtech.com/application-visible-residue-limit-cleaning-valiation?id=&pageID=1&sk=&date= (accessed September 2019)
“Determination of Surface Visible Residue Limits on Pharmaceutical Plant Equipment.” Pharmaceutical Technology 37 (Feb. 2, 2013)
Desai, P., and Walsh, A. “Validation of Visual Inspection As An Analytical Method For Cleaning Validation.” Pharmaceutical Online (Sept. 11, 2017) https://www.pharmaceuticalonline.com/doc/validation-of-visual-inspection-as-an-analytical-method-for-cleaning-validation-0001 (accessed September 2019)
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for human use, Quality Risk Management Q9, (2005)
Walsh, A., et al., “Justification & Qualification Of Visual Inspection For Cleaning Validation In A Low-Risk, Multiproduct Facility.” Pharmaceutical Online (Aug. 3, 2018) https://www.pharmaceuticalonline.com/doc/justification-qualification-of-visual-inspection-for-cleaning-validation-in-a-low-risk-multiproduct-facility-0001 (accessed January 2020)
Forsyth, R.J., and Hartman, J., “A Risk-based Approach to Cleaning Validation using Visible Residue Limits.” Pharmaceutical Engineering 28 (2008) 8–22.
Verweise Teil 2
El Azab W. and Cousin S., “Visual Inspection Practices of Cleaned Equipment: Part I,” accessed April 2020: https://www.pda.org/pda-letter-portal/home/full-article/visual-inspection-practices-of-cleaned-equipment-part-i
Walsh, A., et al., “Justification & Qualification of Visual Inspection for Cleaning Validation in a Low-Risk, Multiproduct Facility.” Pharmaceutical Online (Aug. 3, 2018) https://www.pharmaceuticalonline.com/doc/justification-qualification-of-visual-inspection-for-cleaning-validation-in-a-low-risk-multiproduct-facility-0001
Desai, P. and Walsh, A. “Validation of Visual Inspection as an Analytical Method for Cleaning Validation.” Pharmaceutical Online (Sept. 11, 2017) https://www.pharmaceuticalonline.com/doc/validation-of-visual-inspection-as-an-analytical-method-for-cleaning-validation-0001
Forsyth, R.J., et al. “Visible-Residue Limit for Cleaning Validation and its Potential Application in a Pharmaceutical Research Facility.” Pharmaceutical Technology 28 (Oct. 2004): 58–72 https://pdfs.semanticscholar.org/ea64/d01db84d3b8a5a62f1c824abf8484af363dd.pdf
“Application of Visible-Residue Limit for Cleaning Validation.” Pharmaceutical Technology 29 (Oct. 2, 2005) http://www.pharmtech.com/application-visible-residue-limit-cleaning-validation?id=&pageID=1&sk=&date=
“Determination of Surface Visible Residue Limits on Pharmaceutical Plant Equipment,” Pharmaceutical Technology (Feb. 2, 2013) 37 http://www.pharmtech.com/determination-surface-visible-residue-limits-pharmaceutical-plant-equipment?id=&pageID=1&sk=&date=
Vorgeschlagene Inhalte
Artikel
Ein begründeter Prozess zur Reinigung und Desinfektion
Gesetzliche Leitlinien und Normen, wie der Anhang 1 der Europäischen Guten Herstellungspraxis (EU-GMP), wurden geschaffen, um Standards für Anforderungen zu schaffen. Dies kann zwar hilfreich sein, um bewährte Verfahren zu erreichen, es ist jedoch auch wichtig, sich vor Augen zu halten, dass Verfahren auf die jeweiligen Einrichtungen und Prozesse zugeschnitten sein müssen. In diesem Artikel befasst sich der technische Experte von STERIS, Walid El Azab, mit dem wissenschaftlichen Ansatz, um zu bestimmen, ob vor der Desinfektion ein separater Reinigungsschritt erforderlich ist.
Dienstleistungen, die Sie bei der Validierung Ihrer Geräte berücksichtigen sollten
Die Validierung ist ein wesentlicher Bestandteil Ihres Betriebs. Stellen Sie mithilfe der Validierungsdienste von STERIS sicher, dass Ihre Geräte Ihren Standards entsprechen und die Einhaltung der Branchenvorschriften gewährleistet ist. Unabhängig davon, ob Sie Ihre Ausrüstung zum ersten Mal qualifizieren oder eine jährliche Requalifizierung durchführen: STERIS bietet ein Servicepaket, das Ihren Anforderungen in jedem Schritt des Prozesses gerecht wird.
Weitere Einzelheiten zu den verfügbaren Servicepaketen finden Sie in der Infografik.
Dieses Whitepaper untersucht bewährte Verfahren zur Etablierung und Bewertung von Edelstahlkonservierungsverfahren und Rouge-Minderung in der pharmazeutischen Produktion.
Ein begründeter Prozess zur Reinigung und Desinfektion
Gesetzliche Leitlinien und Normen, wie der Anhang 1 der Europäischen Guten Herstellungspraxis (EU-GMP), wurden geschaffen, um Standards für Anforderungen zu schaffen. Dies kann zwar hilfreich sein, um bewährte Verfahren zu erreichen, es ist jedoch auch wichtig, sich vor Augen zu halten, dass Verfahren auf die jeweiligen Einrichtungen und Prozesse zugeschnitten sein müssen. In diesem Artikel befasst sich der technische Experte von STERIS, Walid El Azab, mit dem wissenschaftlichen Ansatz, um zu bestimmen, ob vor der Desinfektion ein separater Reinigungsschritt erforderlich ist.
Dienstleistungen, die Sie bei der Validierung Ihrer Geräte berücksichtigen sollten
Die Validierung ist ein wesentlicher Bestandteil Ihres Betriebs. Stellen Sie mithilfe der Validierungsdienste von STERIS sicher, dass Ihre Geräte Ihren Standards entsprechen und die Einhaltung der Branchenvorschriften gewährleistet ist. Unabhängig davon, ob Sie Ihre Ausrüstung zum ersten Mal qualifizieren oder eine jährliche Requalifizierung durchführen: STERIS bietet ein Servicepaket, das Ihren Anforderungen in jedem Schritt des Prozesses gerecht wird.
Weitere Einzelheiten zu den verfügbaren Servicepaketen finden Sie in der Infografik.
Dieses Whitepaper untersucht bewährte Verfahren zur Etablierung und Bewertung von Edelstahlkonservierungsverfahren und Rouge-Minderung in der pharmazeutischen Produktion.